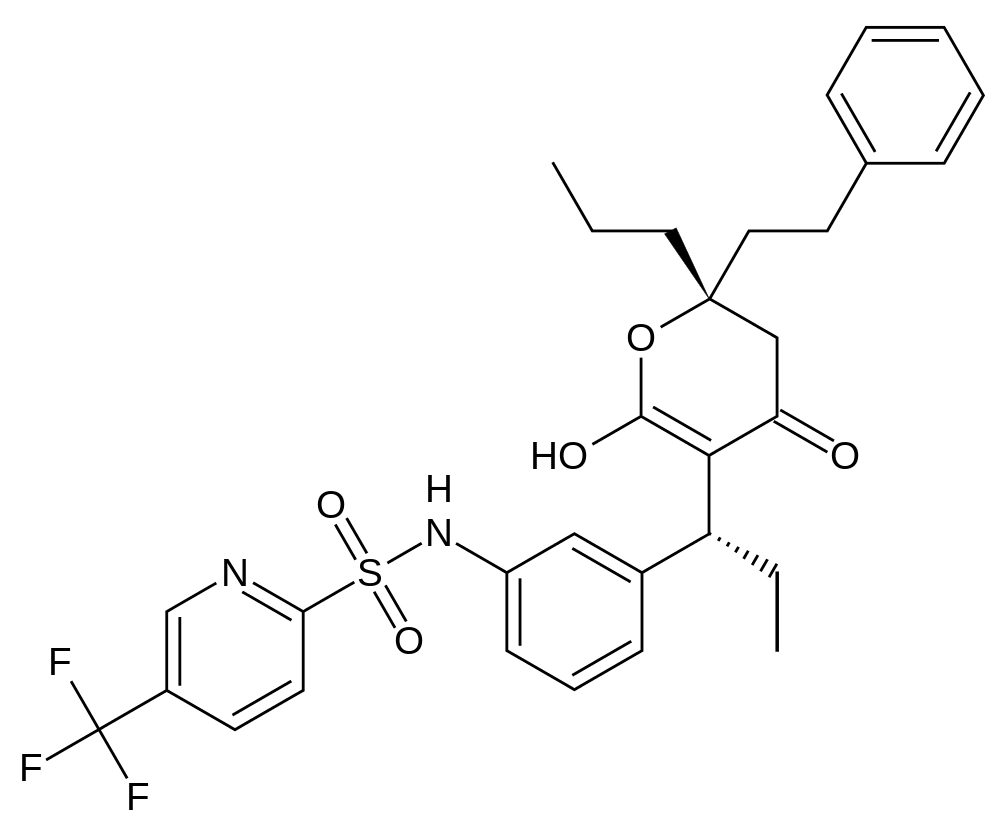
Tipranavir - le virus de l'immunodéficience humaine (vih-1) code pour une aspartyl protéase essentielle pour le clivage et la maturation du précurseur des protéines virales.
Le virus de l'immunodéficience humaine (VIH-1) code pour une aspartyl protéase essentielle pour le clivage et la maturation du précurseur des protéines virales. Le tipranavir est un inhibiteur non peptidique de la protéase du VIH-1, qui inhibe la réplication virale en empêchant la maturation des particules virales.
Activité antivirale in vitro :Le tipranavir inhibe la réplication des souches de laboratoire de VIH-1 et des isolats cliniques dans les modèles aigus d'infection des lymphocytes T, avec des concentrations efficaces à 50 % (CE50) et à 90 % (CE90) comprises entre 0,03 - 0,07 µM (18 à 42 ng/ml) et 0,07 - 0,18 µM (42 à 108 ng/ml), respectivement. Le tipranavir a démontré une activité antivirale in vitro contre un large panel d'isolats du VIH-1 du groupe M sous type non-B (A, C, D, F, G, H, CRF01 AE, CRF02 AG, CRF12 BF). Les isolats du groupe O et du VIH-2 ont, in vitro, une sensibilité réduite au tipranavir, avec des CE50 comprises entre 0,164-1 µM et 0,233-0,522 µM, respectivement. Les études de liaison aux protéines ont montré une diminution de l'activité antivirale du tipranavir, d'un facteur de 3,75 en moyenne, en présence de sérum humain.
Résistance :L'apparition de résistance in vitro au tipranavir est un phénomène lent et complexe. En particulier, lors d'une étude de résistance in vitro, un isolat du VIH-1 ayant une résistance au tipranavir multipliée par un facteur 87 a été sélectionné au bout de 9 mois et présentait 10 mutations au niveau de la protéase (L10F, I13V, V32I, L33F, M36I, K45I, I54V/T, A71V, V82L et I84V), ainsi qu'une mutation au niveau du site de clivage de la polyprotéine gag CA/P2. Des études de génétique inverse ont montré que la présence d'au moins 6 mutations du gène de la protéase (I13V, V32I, L33F, K45I, V82L, I84V) était nécessaire pour conférer une résistance au tipranavir multipliée par un facteur > 10, alors qu'un génotype présentant la totalité des 10 mutations conférait une résistance au tipranavir multipliée par un facteur 69. In vitro, il existe une corrélation inverse entre le degré de résistance au tipranavir et la capacité de réplication des virus. Des virus recombinants, montrant une résistance au tipranavir multipliée par un facteur ≥ 3, ont un taux de réplication inférieur à 1 % par rapport à celui détecté dans les mêmes conditions, chez le VIH-1 de type sauvage. L'émergence in vitro de virus résistants au tipranavir, à partir du VIH-1 de type sauvage, a montré une diminution de la sensibilité aux inhibiteurs de protéase suivants, l'amprénavir, l'atazanavir, l'indinavir, le lopinavir, le nelfinavir et le ritonavir, la sensibilité au saquinavir étant maintenue.Une série d'analyses de régression multivariée regroupant toutes les études cliniques, portant sur les génotypes déterminés à l'inclusion et au cours du traitement, a montré que 16 acides aminés ont été associés à la diminution de la sensibilité au tipranavir et/ou à une diminution de la réponse virologique au bout de 48 semaines : 10V, 13V, 20M/R/V, 33F, 35G, 36I, 43T, 46L, 47V, 54A/M/V, 58E, 69K, 74P, 82L/T, 83D et 84V. Les isolats cliniques ayant une sensibilité au tipranavir diminuée d'un facteur ≥ 10, présentaient des mutations associées au tipranavir au nombre de 8 ou plus. Lors des essais cliniques de phase II et de phase III, les génotypes obtenus en cours de traitement chez 276 patients ont démontré que les principales mutations émergentes sous tipranavir sont L33F/I/V, V82T/L et I84V. L'association de ces trois mutations est généralement nécessaire pour une réduction de la sensibilité. Les mutations en position 82 surviennent selon deux mécanismes : l'un par la mutation 82A préexistante qui sélectionne la mutation 82T et l'autre par un virus de type sauvage présentant la mutation 82V qui sélectionne la mutation 82L.
Résistance croisée : Le tipranavir maintient une activité antivirale significative (résistance 4 fois plus basse) sur la majorité des isolats cliniques du VIH-1, ayant montré après traitement une diminution de la sensibilité aux inhibiteurs de protéase actuellement disponibles, l'amprénavir, l'atazanavir, l'indinavir, le lopinavir, le ritonavir, le nelfinavir et le saquinavir. Chez les virus provenant de patients lourdement prétraités et ayant reçu de nombreux inhibiteurs de protéase peptidiques, il est peu fréquent (moins de 2,5 % des isolats testés) d'observer une augmentation de la résistance au tipranavir d'un facteur supérieur à 10.
Évaluation de l'ECG :L'effet du tipranavir associé au ritonavir à faible dose sur l'intervalle QTcF a été mesuré lors d'une étude au cours de laquelle 81 sujets sains ont reçus deux fois par jour pendant 2,5 jours les traitements suivants : tipranavir/ritonavir (500/200 mg), tipranavir/ritonavir à une dose suprathérapeutique (750/200 mg) et placebo/ritonavir (-/200 mg). Après ajustement en fonction des valeurs initiales et du placebo, la modification moyenne maximale du QTcF était de 3,2 ms (limite supérieure de l'IC unilatéral à 95 % : 5,6 ms) pour la dose de 500/200 mg et de 8,3 ms (limite supérieure de l'IC unilatéral à 95 % : 10,8 ms) pour la dose suprathérapeutique de 750/200 mg. Ainsi, le tipranavir à une dose thérapeutique associé au ritonavir à faible dose n'a pas prolongé l'intervalle QTc, mais pourrait prolonger l'intervalle QTc à une dose suprathérapeutique.
Données de pharmacodynamie clinique : Capsule : cette indication est basée sur les résultats de deux études de phase III, conduites chez des patients adultes hautement prétraités (ayant reçu précédemment un nombre médian de 12 agents antirétroviraux) et porteurs d'un virus résistant aux inhibiteurs de protéase et sur les résultats d'une étude de phase II évaluant la pharmacocinétique, la sécurité et l'efficacité d'TIPRANAVIR chez des patients adolescents âgés de 12 à 18 ans le plus souvent prétraités.Solution buvable : cette indication est basée sur les résultats d'une étude de phase II évaluant la pharmacocinétique, la sécurité et l'efficacité d'TIPRANAVIR solution buvable chez des enfants âgés de 2 à 12 ans le plus souvent prétraités.Les données cliniques suivantes sont issues des analyses à 48 semaines des essais cliniques en cours (RESIST-1 et RESIST-2), mesurant les effets sur le taux plasmatique d'ARN du VIH et le taux de CD4. RESIST-1 et RESIST-2 sont des essais en cours, randomisés, ouverts, multicentriques, conduits chez des patients VIH positifs prétraités par les trois classes d'antirétroviraux. Ces études évaluent le traitement par 500 mg de tipranavir, coadministré avec le ritonavir à faible dose (200 mg, deux fois par jour), en association avec un traitement de base optimisé (TBO) individuellement et défini pour chaque patient sur la base des tests de résistance génotypique et des antécédents du patient. Le traitement comparateur comportait un IP boosté par le ritonavir (également défini individuellement) en association avec le TBO. L'inhibiteur de protéase boosté par le ritonavir était sélectionné parmi le saquinavir, l'amprénavir, l'indinavir ou le lopinavir/ritonavir. Tous les patients avaient reçu au préalable au moins deux traitements antirétroviraux contenant un IP et étaient en échec d'un traitement contenant un IP au moment de l'entrée dans l'étude. A l'inclusion, au moins une mutation primaire au niveau du gène de la protéase parmi les mutations 30N, 46I, 46L, 48V, 50V, 82A, 82F, 82L, 82T, 84V ou 90M devait être présente, et pas plus de deux mutations au niveau des codons 33, 82, 84 ou 90.Après la 8e semaine, les patients du bras comparateur présentant une réponse virologique initiale insuffisante, critère défini par le protocole, avaient la possibilité d'arrêter le traitement et de changer pour le tipranavir associé au ritonavir dans une étude de suivi à long terme.Les 1483 patients inclus dans l'analyse primaire avaient une moyenne d'âge de 43 ans (intervalle : 17 à 80 ans), étaient des hommes à 86 %, étaient blancs à 75 %, noirs à 13 % et asiatiques à 1 %. Dans les bras tipranavir et comparateurs, les taux médians de CD4 à l'inclusion étaient de 158 cellules/mm3 et de 166 cellules/mm3, respectivement (intervalles : 1-1893 et 1-1184 cellules/mm3), les taux plasmatiques médians d'ARN du VIH-1 à l'inclusion étaient de 4,79 log10 copies/ml et de 4,80 log10 copies/ml, respectivement (intervalles : 2,34 - 6,52 et 2,01 - 6,76 log10 copies/ml).Auparavant, les patients avaient été traités en médiane par 6 INTI, 1 INNTI et 4 IP. Dans les 2 études, un total de 67 % de virus des patients étaient résistants et 22 % étaient potentiellement résistants à l'IP comparateur présélectionné. Au total, 10 % des patients avaient été préalablement traités par enfuvirtide. A l'inclusion, les patients présentaient des isolats du VIH-1 avec une médiane de 16 mutations sur le gène de la protéase, et une médiane de 3 mutations primaires du gène de la protéase parmi D30N, L33F/I, V46I/L, G48V, I50V, V82A/F/T/L, I84V et L90M. Concernant les mutations au niveau des codons 33, 82, 84 et 90, environ 4 % des patients ne présentaient aucune de ces mutations, 24 % avaient des mutations au niveau des codons 82 (moins de 1 % avaient la mutation V82L) et 90, 18 % avaient des mutations au niveau des codons 84 et 90 et 53 % avaient au moins une mutation clé au niveau du codon 90. Un patient dans le bras tipranavir présentait les 4 mutations. De plus, la majorité des participants avaient des mutations associées à la résistance aux INTI et aux INNTI. La sensibilité phénotypique à l'inclusion a été évaluée par des prélèvements à l'inclusion chez 454 patients. Il y avait une diminution moyenne de la sensibilité des virus par rapport au virus sauvage d'un facteur 2 pour le tipranavir, d'un facteur 12 pour l'amprénavir, d'un facteur 55 pour l'atazanavir, d'un facteur 41 pour l'indinavir, d'un facteur 87 pour le lopinavir, d'un facteur 41 pour le nelfinavir, d'un facteur 195 pour le ritonavir et d'un facteur 20 pour le saquinavir.A 48 semaines, le critère de réponse combinée au traitement (critère composite défini par le pourcentage de patients présentant une diminution confirmée ≥ 1 log de l'ARN du VIH-1 par rapport à l'inclusion et sans manifestation d'échec au traitement) pour les 2 études était de 34 % dans le bras tipranavir associé au ritonavir et de 15 % dans le bras comparateur. La réponse au traitement est présentée dans le tableau ci-dessous pour l'ensemble de la population (selon l'utilisation d'enfuvirtide ou non), et est détaillée par strate d'IP pour le sous-groupe de patients ayant génotypiquement des souches résistantes.
Pharmacocinétique
Pharmacocinétique - TIPRANAVIR - voie orale
Afin d'obtenir des concentrations plasmatiques efficaces de tipranavir et un traitement quotidien en deux prises par jour, la coadministration de tipranavir avec le ritonavir à faible dose deux fois par jour est indispensable . Le ritonavir agit par inhibition de l'isoenzyme CYP3A4 du cytochrome P450 hépatique, de la pompe d'efflux P-glycoprotéine (P-gp) intestinale et peut-être également de l'isoenzyme CYP3A4 du cytochrome P450 intestinal. Comme il a été démontré lors d'une étude d'escalade de doses conduite chez 113 volontaires sains hommes et femmes VIH-négatifs, le ritonavir augmente l'ASC0-12h, la Cmax et la Cmin et diminue la clairance du tipranavir. La dose de 500 mg de tipranavir, coadministrée avec le ritonavir à faible dose (200 mg ; deux fois par jour), a été associée à une augmentation de 29 fois la moyenne géométrique des taux plasmatiques résiduels matinaux moyens à l'état d'équilibre, par rapport au tipranavir 500 mg deux fois par jour, sans le ritonavir.
Absorption : L'absorption du tipranavir chez l'homme est limitée bien qu'elle n'ait pas encore été quantifiée de façon absolue. Le tipranavir est un substrat de la P-gp, un faible inhibiteur de la P-gp et semble être également un puissant inducteur de la P-gp. Des données suggèrent que, bien que le ritonavir soit un inhibiteur de la P-gp, l'effet résultant d'TIPRANAVIR , coadministré avec le ritonavir à faible dose, à la dose de traitement proposée à l'état d'équilibre, est l'induction de la P-gp. Les concentrations plasmatiques maximales sont atteintes 1 à 5 heures après l'administration de la dose, selon la posologie utilisée. Avec des doses répétées, les concentrations plasmatiques du tipranavir sont inférieures à celles prédites par les données obtenues lors d'une administration unique, probablement en raison de l'induction des enzymes hépatiques. L'état d'équilibre est atteint chez la plupart des sujets après 7 jours de traitement. La pharmacocinétique du tipranavir, coadministré avec du ritonavir à faible dose, est linéaire à l'état d'équilibre.L'administration concomitante de 500 mg d'TIPRANAVIR capsules molles deux fois par jour et de 200 mg de ritonavir 2 fois par jour pendant 2 à 4 semaines, sans restriction alimentaire, a entraîné une concentration plasmatique maximale de tipranavir (Cmax) moyenne de 94,8 ± 22,8 µM chez les femmes (n = 14) et de 77,6 ± 16,6 µM chez les hommes (n = 106), apparaissant environ 3 heures après l'administration. La concentration plasmatique résiduelle moyenne à l'état d'équilibre avant la prise du matin était de 41,6 ± 24,3 µM chez les femmes et de 35,6 ± 16,7 µM chez les hommes. L'ASC du tipranavir après un intervalle de 12 heures suivant l'administration a été en moyenne de 851 ± 309 µM × h (CL = 1,15 l/h) chez les femmes, et de 710 ± 207 µM × h (CL = 1,27 l/h) chez les hommes. La demi-vie moyenne a été de 5,5 heures (chez les femmes) ou 6,0 heures (chez les hommes).
Effets de la nourriture sur l'absorption orale : La nourriture améliore la tolérance du tipranavir associé au ritonavir. Par conséquent, TIPRANAVIR , coadministré avec le ritonavir à faible dose, doit être administré en présence de nourriture.L'absorption du tipranavir, coadministré avec le ritonavir à faible dose, est diminuée en présence d'antiacides .
Distribution : Le tipranavir est fortement lié aux protéines plasmatiques (> 99,9 %). D'après les prélèvements cliniques obtenus chez des volontaires sains et des patients VIH-1-positifs ayant reçu le tipranavir sans le ritonavir, la fraction plasmatique moyenne de tipranavir non lié a été comparable dans les deux populations (volontaires sains : 0,015 % ± 0,006 ; sujets VIH-positifs : 0,019 % ± 0,076). Les concentrations plasmatiques totales de tipranavir de ces échantillons étaient comprises entre 9 et 82 µM. La fraction non liée de tipranavir semblait indépendante de la concentration totale du médicament dans cette zone de concentrations.Aucune étude n'a été effectuée chez l'homme pour déterminer la distribution du tipranavir dans le LCR ou le sperme.
Biotransformation : Les études de métabolisme in vitro effectuées sur des microsomes hépatiques humains ont indiqué que l'isoenzyme CYP3A4 constitue l'isoforme CYP prédominante impliquée dans le métabolisme du tipranavir.La clairance orale du tipranavir a diminué après l'addition de ritonavir, ce qui peut correspondre à une diminution de la clairance de premier passage de la substance au niveau gastro-intestinal et/ou hépatique.En présence de ritonavir à faible dose, le métabolisme du tipranavir est minime. Lors d'une étude clinique menée avec du tipranavir marqué au 14C (500 mg de 14C-tipranavir avec 200 mg de ritonavir, deux fois par jour), le tipranavir inchangé a été prédominant et a représenté 98,4 % ou plus de la radioactivité plasmatique circulante totale 3, 8 ou 12 heures après l'administration. Seuls quelques métabolites ont été retrouvés dans le plasma, tous à l'état de traces (≤ 0,2 % de la radioactivité plasmatique). Le tipranavir non métabolisé a également constitué la majorité de la radioactivité retrouvée dans les fèces (79,9 % de la radioactivité fécale). Dans les fèces, le métabolite fécal le plus abondant, correspondant à 4,9 % de la radioactivité retrouvée dans les selles (soit 3,2 % de la dose administrée), était un métabolite hydroxylé du tipranavir. Dans l'urine, le tipranavir inchangé n'a été retrouvé qu'à l'état de traces (0,5 % de la radioactivité urinaire). Le métabolite urinaire le plus abondant, correspondant à 11 % de la radioactivité retrouvée dans l'urine (soit 0,5 % de la dose administrée) était un glucuroconjugué du tipranavir.
Élimination : L'administration du tipranavir marqué au 14C chez des sujets (n = 8) ayant reçu l'association 500 mg de tipranavir avec 200 mg de ritonavir deux fois par jour à l'état d'équilibre, a démontré que la majorité de la radioactivité (médiane 82,3 %) se retrouvait au niveau des fèces, alors que seulement une médiane de 4,4 % de la dose radioactive était retrouvée dans l'urine. De plus, la majeure partie de la radioactivité (56 %) était excrétée entre 24 et 96 heures après l'administration. La demi-vie moyenne d'élimination efficace de l'association tipranavir avec ritonavir (500 mg/200 mg deux fois par jour avec un repas léger) chez des volontaires sains (n = 67) et des patients adultes infectés par le VIH (n = 120) a été approximativement de 4,8 et 6,0 heures respectivement, à l'état d'équilibre.
Populations particulières : Bien que les données actuellement disponibles soient limitées pour permettre une analyse définitive à ce stade, elles suggèrent que le profil pharmacocinétique est inchangé chez les sujets âgés et est comparable entre les races. Par opposition, l'évaluation à l'état d'équilibre des concentrations plasmatiques résiduelles du tipranavir 10-14 heures après la prise, dans les essais RESIST-1 et RESIST-2, démontre que les femmes ont généralement des concentrations de tipranavir plus élevées que les hommes. Après 4 semaines de traitement par TIPRANAVIR 500 mg associé à 200 mg de ritonavir (2 fois par jour), la concentration plasmatique résiduelle médiane de tipranavir était de 43,9 µM chez les femmes et de 31,1 µM chez les hommes. Cette différence des concentrations ne nécessite pas d'ajustement posologique.
Insuffisance rénale : Les paramètres pharmacocinétiques du tipranavir n'ont pas été étudiés chez les patients présentant une insuffisance rénale. Cependant, la clairance rénale du tipranavir étant négligeable, une diminution de la clairance corporelle totale n'est pas attendue chez les patients présentant une insuffisance rénale.
Insuffisance hépatique : Dans une étude comparant 9 patients atteints d'insuffisance hépatique légère (Child-Pugh classe A) et 9 témoins, l'exposition au tipranavir et au ritonavir, administrés en dose unique et en doses répétées, a augmenté chez les insuffisants hépatiques, tout en restant dans l'intervalle observé au cours des études cliniques. Aucun ajustement posologique n'est nécessaire en cas d'insuffisance hépatique légère, mais les patients doivent être étroitement suivis .L'influence d'une insuffisance hépatique modérée (Child-Pugh classe B) ou sévère (Child-Pugh classe C) sur la pharmacocinétique du tipranavir ou du ritonavir, après administration en doses répétées, n'a pas encore été étudiée. Le tipranavir est contre-indiqué en cas d'insuffisance hépatique modérée ou sévère .
Population pédiatrique : Il a été démontré que la solution buvable a une biodisponibilité supérieure à celle de la forme capsule molle.
Indications
Indications - TIPRANAVIR - usage systémique
TIPRANAVIR , coadministré avec le ritonavir à faible dose, est indiqué dans le traitement de l'infection par le VIH-1 en association avec d'autres agents antirétroviraux, chez les adultes et les adolescents âgés de 12 ans et plus (capsule) et chez les enfants de 2 à 12 ans (solution buvable), lourdement prétraités ayant des virus multirésistants aux inhibiteurs de protéase. TIPRANAVIR doit être utilisé uniquement dans le cadre d'une association de traitements antirétroviraux chez des patients n'ayant pas d'autres alternatives thérapeutiques .Lors de l'instauration du traitement par TIPRANAVIR , coadministré avec le ritonavir à faible dose, les antécédents thérapeutiques de chaque patient et l'analyse des profils de mutations associés aux différents antirétroviraux devront être évalués avec attention. Les tests de résistance génotypique et phénotypique (lorsqu'ils sont disponibles) et les antécédents thérapeutiques doivent guider l'utilisation d'TIPRANAVIR . Lors de l'initiation du traitement, les combinaisons de mutations pouvant avoir un impact négatif sur la réponse virologique au traitement par TIPRANAVIR , coadministré avec le ritonavir à faible dose, doivent être prises en compte .
Surdosage
L'expérience de surdosage chez l'homme avec le tipranavir est très limitée. Aucun signe ou symptôme spécifique de surdosage n'est connu. D'une manière générale, le surdosage pourrait se traduire par une augmentation de la fréquence ou de la sévérité des effets indésirables.Il n'y a pas d'antidote connu au tipranavir en cas de surdosage. Le traitement du surdosage doit comporter des mesures générales de surveillance, telles qu'une surveillance des signes vitaux et de l'état clinique du patient. Si cela est indiqué, l'élimination du tipranavir non absorbé doit être obtenue par vomissement ou lavage gastrique. L'administration de charbon activé peut également être utilisée afin d'aider à l'élimination de la substance non absorbée. Le tipranavir étant fortement lié aux protéines plasmatiques, il est peu probable que la dialyse soit efficace pour une élimination significative de ce médicament.
Grossesse/Allaitement
Contraception chez les hommes et les femmes :Il existe une interaction médicamenteuse entre le tipranavir et les contraceptifs oraux. Par conséquent, une autre méthode de contraception efficace et bien tolérée doit être utilisée pendant le traitement .
Grossesse :Il n'existe pas de données suffisamment pertinentes concernant l'utilisation du tipranavir chez la femme enceinte. Des études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction . Le risque potentiel en clinique n'est pas connu. Le tipranavir ne doit être utilisé pendant la grossesse que si le bénéfice attendu justifie le risque potentiel chez le fœtus.
Allaitement :Conformément aux recommandations déconseillant aux femmes infectées par le VIH d'allaiter leurs nourrissons quelles que soient les circonstances et afin d'éviter tout risque de transmission postnatale du VIH, les mères doivent arrêter l'allaitement lors d'un traitement par TIPRANAVIR .
Fertilité; :Il n'existe pas de données cliniques disponibles sur la fécondité pour le tipranavir. Des études précliniques menées avec le tipranavir n'ont montré aucun effet indésirable sur la fécondité
L'accès aux informations contenues dans ce Site est ouvert à tous les professionnels de santé.
Les informations données sur le site le sont à titre purement informatif.
Toutefois l'utilisateur exploite les données du site Medzai.net sous sa seule responsabilité : ces informations ne sauraient être utilisées sans vérification préalable par le l’internaute et Medzai.net ne pourra être tenue pour responsable des conséquences directes ou indirectes pouvant résulter de l'utilisation, la consultation et l'interprétation des informations fournies.

 Français
Français Русский
Русский