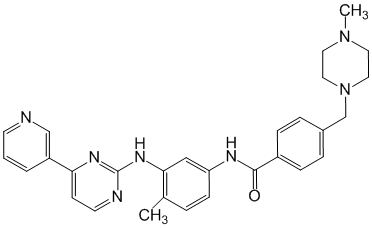
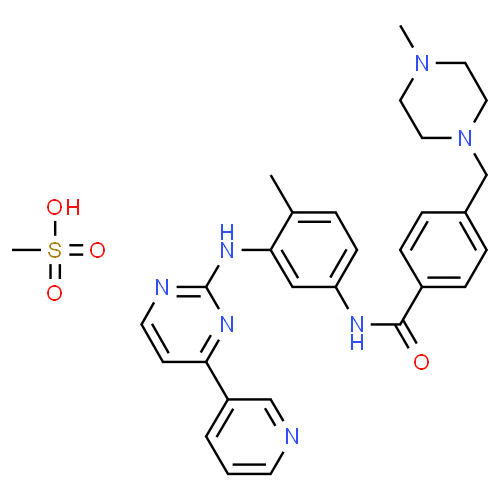
L'imatinib est un inhibiteur de protéine tyrosine kinase qui inhibe puissamment la tyrosine kinase BcrAbl au niveau cellulaire in vitro et in vivo. Le produit inhibe sélectivement la prolifération et induit une apoptose dans les lignées cellulaires Bcr-Abl positives ainsi que dans les cellules leucémiques fraîches provenant de patients atteints de LMC ou de leucémie aiguë lymphoblastique (LAL) chromosome Philadelphie positives.
In vivo, le produit présente une activité anti-tumorale lorsqu'il est administré en monothérapie chez l'animal porteur de cellules tumorales Bcr-Abl positives.
L'imatinib est également un inhibiteur des tyrosine kinases du récepteur du PDGF (platelet-derived growth factor), PDGF-R et du SCF (stem cell factor) c-Kit et il inhibe les processus cellulaires médiés par le PDGF et le SCF. L'activation constitutive du récepteur du PDGF ou des tyrosine kinases Abl, résultant de la fusion à différentes protéines partenaires ou de la production constitutive de PDGF, sont impliquées dans la pathogénèse des SMD/SMP, des SHE/LCE et du DFSP. L'imatinib inhibe la signalisation et la prolifération des cellules sensibles à l'activité dérégulée des kinases Abl ou PDGFR.
Pharmacocinétique
Pharmacocinétique - IMATINIB - voie orale
Paramètres pharmacocinétiques de l'imatinib
La pharmacocinétique de l'imatinib a été évaluée à des doses comprises entre 25 et 1 000 mg. Les profils pharmacocinétiques plasmatiques ont été analysés à J1, puis à J7 ou J28, au moment où les concentrations plasmatiques ont atteint un état d'équilibre.
Absorption
La biodisponibilité absolue moyenne de l'imatinib est de 98%. Il existe une forte variabilité inter-patient de l'ASC de l'imatinib plasmatique après une prise orale. Lorsqu'il est pris au cours d'un repas riche en lipides, le taux d'absorption de l'imatinib est peu réduit (diminution de 11% de la Cmax et prolongation de 1,5 h de tmax), avec une légère diminution de l'ASC (7,4%) comparée à une prise à jeun. L'effet d'une chirurgie gastro-intestinale antérieure sur l'absorption du produit n'a pas été étudié.
Distribution
A des concentrations d'imatinib cliniquement significatives, la fraction liée aux protéines plasmatiques est approximativement de 95%, sur la base des études in vitro ; il s'agit principalement d'une liaison à l'albumine et aux alpha-glycoprotéines acides, et dans une faible mesure aux lipoprotéines.
Biotransformation
Chez l'homme, le principal métabolite circulant est le dérivé pipérazine N-déméthylé qui présente in vitro une activité similaire à l'imatinib. L'ASC plasmatique de ce métabolite n'atteint que 16% de l'ASC de l'imatinib. L'affinité pour les protéines plasmatiques du métabolite N-déméthylé est similaire à celle de la molécule mère.
L'imatinib et le métabolite N-déméthylé représentent au total environ 65% du taux circulant de radioactivité (ASC(0-48h)). Le taux circulant de radioactivité restant correspond à un nombre de métabolites mineurs.
Les tests in vitro montrent que le CYP3A4 est le principal enzyme du cytochrome P450 humain catalysant la biotransformation de l'imatinib. Parmi un éventail de médicaments potentiellement coadministrés (paracétamol, aciclovir, allopurinol, amphotéricine, cytarabine, érythromycine, fluconazole, hydroxyurée, norfloxacine, pénicilline V) seuls l'érythromycine (IC50 50 μM) et le fluconazole (IC50 118 μM) ont montré une inhibition du métabolisme de l'imatinib pouvant être cliniquement significative.
In vitro, l'imatinib est un inhibiteur compétitif des substrats marqués du CYP2C9, des CYP2D6 et des CYP3A4/5 avec des valeurs de Ki de 27, 7,5 et 7, 9 µmol/l respectivement obtenues sur les microsomes hépatiques humains. Les concentrations plasmatiques maximales de l'imatinib sont de 2- 4 μmol/l. Par conséquent, une inhibition du métabolisme de produits co-administrés mettant en jeu les CYP2D6 et CYP3A4/5 est possible. L'imatinib n'interfère pas avec la biotransformation du 5- fluorouracile mais inhibe le métabolisme du paclitaxel par inhibition compétitive du CYP2C8 (Ki = 34,7 µM). Cette valeur de Ki est de loin supérieure aux taux plasmatiques d'imatinib prévisibles chez les patients. Par conséquent, aucune interaction n'est attendue en cas de co-administration de l'imatinib avec le 5-fluorouracile ou le paclitaxel.
Élimination
Après administration d'une dose orale d'imatinib marqué au 14C, environ 81% de la dose est éliminée au bout de 7 jours (68% dans les fèces et 13% dans les urines). La forme inchangée représente 25% de la dose (5% dans les urines, 20% dans les fèces), le reste étant composé de métabolites.
Pharmacocinétique plasmatique
Après administration par voie orale chez le volontaire sain, la demi-vie, d'environ 18 h, est compatible avec une prise quotidienne unique. L'augmentation de l'ASC moyenne de l'imatinib est linéaire et proportionnelle à la dose administrée à des doses orales allant de 25 à 1 000 mg. Lors 25 d'administrations répétées en prise quotidienne unique, la cinétique de l'imatinib n'est pas modifiée, mais son accumulation, à l'état d'équilibre, est augmentée d'un facteur de 1,5 à 2,5.
Pharmacocinétiques de population
Une analyse de pharmacocinétique de population de patients atteints de LMC a montré une légère influence de l'âge sur le volume de distribution (augmentation de 12% chez les patients > 65 ans), mais cette variation ne semble pas cliniquement significative. Bien que l'effet du poids corporel sur la clairance de l'imatinib laisse attendre une clairance moyenne de 8,5 l/h pour un patient pesant 50 kg, contre 11,8 l/h pour un patient pesant 100 kg, une adaptation de la posologie en fonction du poids n'est pas requise. Le sexe n'a aucune influence sur les paramètres cinétiques de l'imatinib.
Pharmacocinétique chez l'enfant
Comme chez l'adulte, l'imatinib a été rapidement absorbé après administration orale chez le patient pédiatrique dans des études de phase I et de phase II. Chez l'enfant, l'administration de doses de 260 et 340 mg/m²/jour a permis d'obtenir des concentrations plasmatiques équivalentes aux doses de respectivement 400 mg et 600 mg chez l'adulte. La comparaison de l'ASC(0-24) à J8 et J1 pour une dose de 340 mg/m²/jour a révélé une accumulation de 1,7 fois après des prises uniques quotidiennes itératives.
Des analyses poolées de données de pharmacocinétique de population chez les enfants atteints d'affections hématologiques (LMC, LAL Ph+, ou autres affections hématologiques traitées par l'imatinib) ont montré que la clairance de l'imatinib augmente parallèlement à celle de la surface corporelle (SC). Après correction de l'effet de la SC, d'autres caractéristiques démographiques telles que l'âge, le poids corporel, et l'indice de masse corporelle n'avaient pas d'effet cliniquement significatif sur l'exposition à l'imatinib. L'analyse a confirmé que l'exposition à l'imatinib chez les enfants recevant 260 mg/m² une fois par jour (sans dépasser 400 mg une fois par jour) ou 340 mg/m² une fois par jour (sans dépasser 600 mg une fois par jour) était comparable à celle des adultes qui ont reçu 400 mg ou 600 mg d'imatinib une fois par jour.
Altération des fonctions organiques
L'imatinib et ses métabolites ne sont pas excrétés de façon significative par le rein. Les patients ayant une altération de la fonction rénale légère à modérée présentent une exposition plasmatique supérieure à celle des patients présentant une fonction rénale normale. L'augmentation est approximativement 1,5 à 2 fois plus, correspondant à une augmentation de 1,5 fois le taux plasmatique d'AGP à laquelle l'imatinib est fortement lié. La clairance de l'imatinib libre chez les patients ayant une altération de la fonction rénale est probablement similaire à celle des patients avec une fonction rénale normale, puisque l'excrétion rénale représente une voie d'élimination mineure de l'imatinib .
Dosage
Dosage - IMATINIB - voie orale
Comprimé pelliculé
Le traitement doit être instauré par un médecin ayant l'expérience du traitement des hémopathies malignes ou des sarcomes.
Pour les doses de 400 mg et plus (voir les recommandations de doses ci-dessous), un comprimé de 100 mg est disponible.
La dose prescrite doit être administrée par voie orale avec un grand verre d'eau, au cours d'un repas pour réduire le risque d'irritations gastro-intestinales. Les doses de 400 mg ou 600 mg devront être administrées en une prise par jour, tandis que la dose journalière de 800 mg devra être répartie en deux prises de 400 mg par jour, matin et soir.
Pour les patients incapables d'avaler les comprimés pelliculés, il est possible de disperser ces comprimés dans un verre d'eau plate ou de jus de pomme. Le nombre de comprimés requis devra être placé dans un volume de boisson approprié (approximativement 50 ml pour un comprimé à 100 mg et 200 ml pour un comprimé à 400 mg) et être remué avec une cuillère. La suspension devra être administrée immédiatement après désagrégation complète du (des) comprimé(s).
Posologie dans la LMC chez l'adulte
Chez les patients adultes en phase chronique de LMC, la posologie recommandée d'imatinib est de 400 mg/j. La phase chronique est définie par l'ensemble des critères suivants : blastes < 15% dans le sang et la moelle osseuse, basophiles dans le sang < 20%, plaquettes > 100 x 109/l.
Chez les patients adultes en phase accélérée, la posologie recommandée d'imatinib est de 600 mg/j. La phase accélérée est définie par la présence d'un des critères suivants : blastes ≥ 15% mais < 30% dans le sang ou la moelle osseuse, blastes plus promyélocytes ≥ 30% dans le sang ou la moelle osseuse (à condition que blastes < 30%), basophiles dans le sang ≥ 20%, plaquettes < 100 x 109/l indépendamment du traitement.
Chez les patients adultes en crise blastique, la posologie recommandée est de 600 mg/j d'IMATINIB. La crise blastique est définie par la présence de blastes ≥ 30% dans le sang ou la moelle osseuse ou un envahissement extramédullaire autre qu'une hépatosplénomégalie.
Durée du traitement : dans les études cliniques, le traitement par l'imatinib est poursuivi jusqu'à la progression de la maladie. L'effet de l'arrêt du traitement après l'obtention d'une réponse cytogénétique complète n'a pas été étudié.
En l'absence d'effets indésirables sévères et de neutropénie ou de thrombopénie sévères non imputables à la leucémie, une augmentation de la dose peut être envisagée de 400 mg à 600 mg ou 800 mg, chez les patients en phase chronique, ou de 600 mg à un maximum de 800 mg (en deux prises de 400 mg par jour) chez les patients en phase accélérée ou en crise blastique, dans les circonstances suivantes : évolution de la maladie (à tout moment), absence de réponse hématologique satisfaisante après un minimum de 3 mois de traitement, absence de réponse cytogénétique après 12 mois de traitement, ou perte de la réponse hématologique et/ou cytogénétique obtenue auparavant.
Les patients devront être surveillés étroitement après augmentation de la dose étant donnée la possibilité d'une incidence accrue des effets indésirables à plus fortes doses.
Posologie dans la LMC chez l'enfant
Chez l'enfant, la posologie devra être établie en fonction de la surface corporelle (mg/m²). La dose journalière recommandée chez l'enfant est de 340 mg/m2 dans la LMC en phase chronique et dans la LMC en phase avancée (ne doit pas dépasser une dose totale de 800 mg). Le traitement peut être administré en une prise quotidienne ou bien être divisé en deux prises (une le matin et une le soir). Ces recommandations posologiques reposent actuellement sur un faible nombre d'enfants .
On ne dispose d'aucune donnée chez l'enfant de moins de 2 ans.
L'augmentation de doses de 340 mg/m² jusqu'à 570 mg/m² par jour (sans dépasser la dose totale de 800 mg) peut être envisagée chez l'enfant en l'absence d'effets indésirables graves et de neutropénie ou thrombopénie sévères non liées à la leucémie dans les circonstances suivantes : progression de la maladie (à n'importe quel moment) ; absence de réponse hématologique satisfaisante après au moins 3 mois de traitement ; absence de réponse cytogénétique après 12 mois de traitement ; ou perte d'une réponse hématologique et/ou cytogénétique antérieure. Les patients devront être surveillés attentivement au cours des escalades de doses compte tenu du risque accru d'effets indésirables à des doses plus élevées.
Posologie dans les LAL Ph+ chez les patients adultes
La posologie recommandée d'IMATINIB est de 600 mg/jour chez les patients adultes atteints de LAL Ph+. Le traitement devrait être supervisé par des hématologues experts dans la prise en charge de cette maladie pour toutes les phases de traitement.
Schéma thérapeutique : sur la base des données existantes, l'imatinib s'est montré efficace et sûr lorsqu'il est administré à 600 mg/j en association à une chimiothérapie d'induction, de consolidation et d'entretien utilisée des LAL Ph+ nouvellement diagnostiquées de l'adulte . La durée du traitement par IMATINIB peut varier en fonction du traitement appliqué, mais généralement les traitements prolongés d'imatinib ont fourni de meilleurs résultats.
Chez les patients adultes atteints de LAL Ph+ en rechute ou réfractaire, une monothérapie par imatinib à la dose de 600 mg/j est sûre, efficace et peut être poursuivie jusqu'à la progression de la maladie.
Posologie dans les LAL Ph+ chez l'enfant
Chez l'enfant, la posologie devra être établie en fonction de la surface corporelle (mg/m2). Dans les LAL Ph+, la dose journalière recommandée chez l'enfant est de 340 mg/m2 (sans dépasser une dose totale de 600 mg).
Posologie dans les SMD/SMP
La posologie recommandée dIMATINIB est de 400 mg/jour chez les patients adultes atteints de SMD/SMP.
Durée du traitement : dans l'unique étude clinique menée à ce jour, le traitement par IMATINIB a été poursuivi jusqu'à la progression de la maladie . A la date de l'analyse, la durée médiane de traitement était de 47 mois (24 jours à 60 mois).
Posologie dans les SHE/LCE
La dose recommandée dIMATINIB est de 100 mg/jour chez les patients adultes atteints de SHE/LCE.
Une augmentation de dose de 100 mg à 400 mg chez ces patients peut être envisagée si la réponse au traitement est insuffisante et en l'absence d'effets indésirables.
Le traitement doit être poursuivi aussi longtemps qu'il est bénéfique pour le patient.
Posologie dans le DFSP
La posologie recommandée dIMATINIB est de 800 mg/jour chez les patients adultes atteints de DFSP.
Ajustement de la posologie en cas d'effets indésirables
Effets indésirables extra-hématologiques
En cas de survenue d'un effet indésirable extra-hématologique sévère, le traitement par IMATINIB doit être interrompu jusqu'à résolution de l'événement. Le traitement peut ensuite être repris de manière appropriée en fonction de la sévérité initiale de l'événement.
En cas d'élévation de la bilirubine > 3 x la limite supérieure de la normale (LSN) fournie par le laboratoire d'analyses ou des transaminases > 5 x la LSN, IMATINIB doit être interrompu jusqu'à un retour de la bilirubine à un taux < 1,5 x la LSN et des transaminases à un taux < 2,5 x la LSN. Le traitement peut alors être repris à dose réduite chez l'adulte, la dose sera diminuée de 400 à 300 mg ou de 600 à 400 mg ou de 800 mg à 600 mg, et chez l'enfant la dose sera diminuée de 340 à 260 mg/m² /jour.
Effets indésirables hématologiques
En cas de neutropénie ou thrombopénie sévères, il est recommandé de diminuer la dose ou d'interrompre le traitement conformément au tableau ci-dessous.
Ajustements de posologie en cas de neutropénie et de thrombocytopénie :
SHE/LCE (dose initiale de 100 mg)
PN < 1,0 x 109/l
et/ou
plaquettes < 50 x 109/l
1. Arrêter IMATINIB jusqu'à ce que PN ≥ 1,5 x 109/l et plaquettes ≥ 75 x 109/l.
2. Reprendre le traitement par IMATINIB à la dose antérieure (c'est à dire avant l'effet indésirable sévère).
LMC en phase chronique,
SMD/SMP (dose initiale 400 mg)
SHE/LCE (à la dose de 400 mg)
PN < 1,0 x 109/l
et/ou
plaquettes < 50 x 109/l
1. Arrêter IMATINIB jusqu'à ce que PN ≥ 1,5 x 109/l et plaquettes ≥ 75 x 109/l.
2. Reprendre le traitement par IMATINIB à la dose antérieure (c'est à dire avant l'effet indésirable sévère).
3. En cas de récidive de PN < 1,0 x 109/l et/ou plaquettes < 50 x 109/l, répéter l'étape 1 puis reprendre IMATINIB à la dose de 300 mg.
LMC en phase chronique en pédiatrie
(à la dose de 340 mg/m2)
PN < 1,0 x 109/l
et/ou
plaquettes < 50 x 109/l
1. Arrêter IMATINIB jusqu'à ce que PN ≥ 1,5 x 109/l et plaquettes ≥ 75 x 109/l.
2. Reprendre le traitement par IMATINIB à la dose antérieure (c'est à dire avant l'effet indésirable sévère).
3. En cas de récidive de PN < 1,0 x 109/l et/ou plaquettes < 50 x 109/l, répéter l'étape 1 puis reprendre IMATINIB à la dose de 260 mg/m2.
LMC en phase accélérée ou crise blastique et LAL Ph+ (dose initiale 600 mg)
aPN < 0,5 x 109/l
et/ou
plaquettes < 10 x 109/l
1. Vérifier si la cytopénie est imputable à la leucémie (ponction ou biopsie médullaire).
2. Si la cytopénie n'est pas imputable à la leucémie, diminuer la dose d'IMATINIB à 400 mg.
3. Si la cytopénie persiste pendant 2 semaines, diminuer encore la dose à 300 mg.
4. Si la cytopénie persiste pendant 4 semaines et n'est toujours pas imputable à la leucémie, arrêter imatinib jusqu'à ce que PN ≥ 1 x 109/l et plaquettes ≥ 20 x 109/l, puis reprendre le traitement à 300 mg.
LMC en phase accélérée ou en crise blastique en pédiatrie
(à la dose de 340 mg/m2)
aPN < 0,5 x 109/l
et/ou
plaquettes < 10 x 109/l
1. Vérifier si la cytopénie est imputable à la leucémie (ponction ou biopsie médullaire).
2. Si la cytopénie n'est pas imputable à la leucémie, diminuer la dose d'IMATINIB à 260 mg/m2.
3. Si la cytopénie persiste pendant 2 semaines, diminuer encore la dose à 200 mg/m2.
4. Si la cytopénie persiste pendant 4 semaines et n'est toujours pas imputable à la leucémie, arrêter IMATINIB jusqu'à ce que PN ≥ 1 x 109/l et plaquettes ≥ 20 x 109/l, puis reprendre le traitement à 200 mg/m2.
DFSP
(à la dose de 800 mg)
PN < 1,0 x 109/l
et/ou
plaquettes < 50 x 109/l
1. Arrêter IMATINIB jusqu'à ce que PN ≥ 1,5 x 109/l et plaquettes ≥ 75 x 109/l.
2. Reprendre le traitement par imatinib à la dose de 600 mg.
3. En cas de récidive de PN < 1,0 x 109/l et/ou plaquettes < 50 x 109/l, répéter l'étape 1 puis reprendre imatinib à la dose réduite de 400 mg.
PN = Polynucléaires Neutrophiles
asurvenant après au moins 1 mois de traitement
Populations spéciales
Enfant : il n'y a pas d'expérience chez l'enfant de moins de 2 ans atteint de LMC et chez l'enfant de moins d'un an atteint de LAL Ph+ . L'expérience est très limitée chez l'enfant atteint de SMD/SMP, de DFSP et de SHE/LCE.
La sécurité et l'efficacité de l'imatinib chez les enfants âgés de moins de 18 ans atteints de SMD/SMP, DFSP et SHE/LCE n'ont pas été établies par des études cliniques.
Insuffisance hépatique : L'imatinib est principalement métabolisé par le foie. Les patients présentant une altération de la fonction hépatique, légère, modérée ou importante devraient être traités à la dose quotidienne minimale recommandée 400 mg. La dose peut être réduite si elle est mal tolérée .
Classification des altérations hépatiques :
Altération de la fonction hépatique
Tests de la fonction hépatique
Légère
Augmentation de la bilirubine totale de 1,5 fois la LSN
ASAT : > LSN (peut être normale ou < LSN si la bilirubine totale est > LSN)
Modérée
Augmentation de la bilirubine totale > à 1,5 fois la LSN et < 3,0 fois la LSN
ASAT quelle que soit la valeur
Importante
Augmentation de la bilirubine totale > 3,0 fois la LSN et < 10 fois la LSN
ASAT quelle que soit la valeur
LSN : Limite supérieure de la normale du laboratoire
ASAT : aspartate aminotransférase
Insuffisance rénale :
Chez les patients présentant une altération de la fonction rénale ou sous dialyse, la dose initiale de traitement de 400 mg par jour est recommandée. Toutefois, la prudence est recommandée chez ces patients. La dose peut être réduite si elle est mal tolérée. Si elle est tolérée, la dose peut être augmentée en l'absence d'efficacité .
Personnes âgées :
La pharmacocinétique de l'imatinib n'a pas été spécifiquement étudiée chez les personnes âgées. Aucune différence significative de pharmacocinétique n'a été observée en fonction de l'âge chez les patients adultes inclus dans les études cliniques dont plus de 20% étaient âgés de 65 ans et plus. Par conséquent, aucune recommandation particulière sur la posologie n'est requise chez les personnes âgées.
Indications
Indications - IMATINIB - usage systémique
IMATINIB est indiqué dans le traitement :
des patients adultes et enfants atteints de leucémie myéloïde chronique (LMC) chromosome Philadelphie (bcr-abl) positive (Ph+) nouvellement diagnostiquée lorsque la greffe de moelle osseuse ne peut être envisagée comme un traitement de première intention.
des patients adultes et enfants atteints de LMC Ph+ en phase chronique après échec du traitement par l'interféron-alpha, ou en phase accélérée ou en crise blastique.
des patients adultes et enfants atteints de leucémie aiguë lymphoïde chromosome Philadelphie positive (LAL Ph+) nouvellement diagnostiquée en association avec la chimiothérapie.
des patients adultes atteints de LAL Ph+ réfractaire ou en rechute en monothérapie.
des patients adultes atteints de syndromes myélodysplasiques/myéloprolifératifs (SMD/SMP) associés à des réarrangements du gène du PDGFR (platelet-derived growth factor receptor).
des patients adultes atteints d'un syndrome hyperéosinophilique (SHE) à un stade avancé et/ou d'une leucémie chronique à éosinophiles (LCE) associés à un réarrangement du FIP1L1- PDGFRα.
L'effet d'imatinib sur l'issue d'une greffe de moelle osseuse n'a pas été évalué.
IMATINIB est indiqué dans :
le traitement des patients adultes atteints de dermatofibrosarcome protuberans (DFSP ou maladie de Darier-Ferrand) non résécable et des patients adultes atteints de DFSP en rechute et/ou métastatique ne relevant pas d'un traitement chirurgical.
Chez l'adulte et les patients pédiatriques, l'efficacité de l'imatinib est basée sur les taux de réponses hématologiques et cytogénétiques globales et la survie sans progression dans la LMC, sur les taux de réponses hématologique et cytogénétique des LAL Ph+, des SMD/SMP, sur les taux de réponses hématologiques des SHE/LCE et sur les taux de réponses objectives des patients adultes dans les DFSP non-résécables et/ou métastatiques. L'expérience avec l'imatinib chez les patients atteints de SMD/SMP associés à des réarrangements du gène du PDG FR est très limitée . A l'exception de la LMC en phase chronique nouvellement diagnostiquée, il n'existe pas d'étude clinique contrôlée démontrant un bénéfice clinique ou une prolongation de la durée de vie, pour ces maladies.
Effets indésirables
Effets indésirables - IMATINIB - usage systémique
Résumé du profil de sécurité
Les patients atteints de pathologies malignes à un stade avancé peuvent présenter des affections intercurrentes. Ces affections peuvent rendre difficile l'évaluation du lien entre l'administration de l'imatinib et la survenue d'événements indésirables en raison de la variété des symptômes liés à la maladie sous-jacente, à sa progression ou à la co-administration de nombreux médicaments.
Au cours des études cliniques menées dans la LMC, un arrêt du traitement motivé par des effets indésirables imputables au médicament a été observé chez 2,4% des patients nouvellement diagnostiqués, 4% des patients en phase chronique tardive après échec du traitement par l'interféron, 4% des patients en phase accélérée après échec du traitement par l'interféron et 5% des patients en crise blastique après échec du traitement par l'interféron.
Les effets indésirables ont été comparables dans toutes les indications. Les effets indésirables les plus fréquemment rapportés (≥ 10%) pouvant être imputables au traitement par l'imatinib ont été des nausées modérées, vomissements, diarrhée, douleur abdominale, fatigue, myalgies, crampes musculaires et rash. Des dèmes superficiels ont été très fréquemment observés dans toutes les études cliniques et décrits principalement comme des dèmes périorbitaux ou des membres inférieurs. Toutefois, ces dèmes ont été rarement sévères et ont pu être contrôlés par des diurétiques, d'autres mesures symptomatiques ou en réduisant la dose d'imatinib.
Lorsque l'imatinib était associé à des doses élevées de chimiothérapie chez des patients atteints de LAL Ph+, une toxicité hépatique transitoire se traduisant par une élévation des transaminases et une hyperbilirubinémie ont été observées. Au vu des données limitées de tolérance, les effets indésirables rapportés ci-après chez l'enfant sont cohérents avec le profil de tolérance observé chez l'adulte atteint de LAL Ph+. Les données de tolérance chez l'enfant atteint de LAL Ph+ sont très limitées bien qu'aucun nouveau problème de sécurité n'ait été identifié.
Divers effets indésirables tels qu'épanchement pleural, ascite, dème pulmonaire, prise de poids rapide avec ou sans dème superficiel ont été décrits dans le cadre de rétention hydrique. Ces effets peuvent habituellement être contrôlés par l'interruption temporaire de l'imatinib et par l'utilisation de diurétiques et d'autres traitements symptomatiques appropriés. Cependant, certains de ces effets peuvent être graves voire mettre en jeu le pronostic vital : plusieurs patients en crise blastique sont décédés, avec un tableau clinique complexe associant un épanchement pleural, une insuffisance cardiaque congestive et une insuffisance rénale. Les études cliniques menées chez l'enfant n'ont pas révélé de données de tolérance particulière à cette population.
Les effets indésirables
Les effets indésirables, en dehors des cas isolés, sont repris ci-dessous par classe de systèmes d'organes et par ordre de fréquence Les catégories de fréquence sont définies selon la convention suivante : très fréquent (≥1/10), fréquent (≥1/100, <1/10), peu fréquent (≥1/1 000, <1/100), rare (≥1/10 000, <1/1 000), très rare (<1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Au sein de chaque groupe de fréquence, les effets indésirables doivent être présentés suivant un ordre de fréquence, le plus fréquent en premier.
Les effets indésirables et leurs fréquences sont présentés dans le Tableau 1.
Tableau 1 - Tableau de synthèse des effets indésirables
Infections et infestations
Peu fréquent
Zona, herpès simplex, inflammation nasopharyngée, pneumonie1, sinusite, cellulites, infection des voies respiratoires hautes, grippe, infection des voies urinaires, gastro-entérite, septicémie
Rare
Infection fongique
Fréquence indéterminée
Réactivation de l'hépatite B
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes)
Rare:
Syndrome de lyse tumorale
Fréquence indéterminée
Hémorragie tumorale/nécrose tumorale*
Affections du système immunitaire
Fréquence indéterminée
Choc anaphylactique*
Affections hématologiques et du système lymphatique
Hypokaliémie, augmentation de l'appétit, hypophosphatémie, diminution de l'appétit, déshydratation, goutte, hyperuricémie, hypercalcémie, hyperglycémie, hyponatrémie
Rare
Hyperkaliémie, hypomagnésémie
Affections psychiatriques
Fréquent
Insomnie
Peu fréquent
Dépression, diminution de libido, anxiété
Rare
Confusion
Affections du système nerveux
Très fréquent
Céphalées
Fréquent
Sensations vertigineuses, paresthésies, troubles du goût, hypoesthésie
Peu fréquent
Migraine, somnolence, syncope, neuropathie périphérique, troubles de la mémoire, sciatique, syndrome des jambes sans repos, tremblement, hémorragie cérébrale
Gynécomastie, dysfonctionnement érectile, ménorragie, menstruation irrégulière, troubles sexuels, douleur des mamelons, gonflement des seins, dème du scrotum
Rare
Corps jaune hémorragique, kyste ovarien hémorragique
Troubles généraux et anomalies au site d'administration
Très fréquent
Rétention hydrique et dème, fatigue
Fréquent
Faiblesse, pyrexie, anasarque, frissons, rigidité
Peu fréquent
Douleur thoracique, malaise
Investigations
Très fréquent
Prise de poids
Fréquent
Perte de poids
Peu fréquent
Augmentation de la créatininémie, augmentation de la créatine phosphokinase, augmentation de la lacticodeshydrogénase, augmentation des phosphatases alcalines
Rare
Augmentation de l'amylasémie
*Les types de réactions suivantes ont principalement été observées au cours de la commercialisation d'imatinib. Cela comprend les rapports spontanés de cas individuels ainsi que les effets indésirables graves des études cliniques en cours, des programmes d'accès élargi et des études de pharmacologie clinique et des études exploratoires menées dans le cadre d'indications thérapeutiques non enregistrées. Comme ces effets sont issus d'une population dont la taille n'est pas déterminée, il n'est pas toujours possible d'estimer de manière fiable leur fréquence ou d'établir la relation de causalité avec l'exposition à l'imatinib.
1 La pneumonie a été le plus fréquemment observée chez les patients atteints de LMC en transformation.
3 Selon l'unité de mesure « patient-année », les effets cardiaques incluant l'insuffisance cardiaque congestive ont été plus fréquemment observés chez les patients ayant une LMC en transformation que chez ceux ayant une LMC en phase chronique.
4 Les saignements (hématomes et hémorragies) ont été le plus fréquemment observés chez les patients atteints de LMC en transformation (LMC en phase accélérée et LMC en crise blastique).
5 L'épanchement pleural a été rapporté plus fréquemment chez les patients ayant une LMC en transformation (LMC en phase accélérée et LMC en crise blastique) que chez les patients en phase chronique.
8 Des cas mortels d'insuffisance hépatique et de nécrose hépatique ont été rapportés.
9 Les douleurs musculosquelettiques et les effets reliés à ces douleurs ont été plus fréquemment observés chez les patients atteints de LMC.
10 Des cas d'évolution fatale ont été rapportés chez des patients à un stade avancé de la maladie, présentant des infections sévères, des neutropénies sévères et d'autres troubles cliniques concomitants sévères.
Anomalies biologiques
Paramètres hématologiques
Dans les LMC, des cytopénies, en particulier des neutropénies et des thrombopénies, ont été régulièrement rapportées dans toutes les études cliniques avec une fréquence plus élevée aux fortes doses ≥ 750 mg (étude de phase I). Cependant, la survenue des cytopénies dépendait aussi nettement du stade de la maladie : la fréquence des neutropénies et des thrombopénies de grade 3 ou 4 (PN < 1,0 x 109 /l ; taux de plaquettes < 50 x 109 /l) étant 4 à 6 fois plus élevée dans les LMC en crise blastique ou en phase accélérée (respectivement 59-64% pour les neutropénies et 44-63% pour les thrombopénies) que dans les LMC en phase chronique nouvellement diagnostiquées (16,7% de neutropénie et 8,9% de thrombopénie). Dans les LMC en phase chronique nouvellement diagnostiquées, les neutropénies et les thrombopénies de grade 4 (PN< 0,5 x 109 /l; plaquettes < 10 x 109 /l) ont été observées chez 3,6% et < 1% des patients respectivement. La durée médiane des épisodes de neutropénie est habituellement de l'ordre de 2 à 3 semaines, et de 3 à 4 semaines pour les épisodes de thrombopénie. Ces événements peuvent être habituellement contrôlés soit par une réduction de la dose soit par une interruption du traitement par l'imatinib, mais peuvent dans de rares cas conduire à une interruption définitive du traitement. En pédiatrie, chez les enfants atteints de LMC, les toxicités les plus fréquemment observées étaient des cytopénies de grade 3 ou 4 incluant des neutropénies, des thrombocytopénies et des anémies. Elles surviennent principalement dans les premiers mois de traitement.
Paramètres biochimiques
Des augmentations importantes des transaminases (< 5%) ou de la bilirubine (< 1%) ont été observées chez des patients atteints de LMC et ont été habituellement contrôlées par une réduction de la dose ou une interruption du traitement (la durée médiane de ces épisodes était d'environ une semaine). Le traitement a été interrompu définitivement en raison d'anomalies biologiques hépatiques chez moins de 1% des patients atteints de LMC.
Il y a eu des cas d'hépatite cytolytique et cholestatique et de défaillance hépatique. Dans certains cas l'issue fut fatale, notamment pour un patient sous dose élevée de paracétamol.
Description de certains effets indésirables
Réactivation de l'hépatite B
Des cas de réactivation du virus de l'hépatite B ont été rapportés chez des patients traités par des inhibiteurs de la tyrosine kinase BCR-ABL. Certains de ces cas ont évolué vers une insuffisance hépatique aiguë ou une hépatite fulminante requérant une transplantation hépatique ou dont l'issue a été fatale .
Grossesse/Allaitement
Femmes en âge de procréer
Les femmes en âge de procréer doivent être informées qu'elles doivent utiliser une contraception efficace pendant le traitement.
Grossesse
Les données concernant l'utilisation de l'imatinib chez la femme enceinte sont limitées. Depuis la mise sur le marché, des cas d'avortement spontanés et d'anomalies congénitales ont été rapportés chez des femmes ayant été traitées par imatinib. Les études effectuées chez l'animal ont toutefois mis en évidence une toxicité sur la reproduction et le risque potentiel sur le ftus en clinique n'est pas connu. L'imatinib ne doit pas être utilisé pendant la grossesse à moins d'une nécessité absolue. S'il est utilisé au cours de la grossesse, la patiente doit être prévenue du risque potentiel pour le ftus.
Allaitement
Les informations concernant la distribution de l'imatinib dans le lait maternel sont limitées. Les études chez deux patientes qui allaitaient ont montré que l'imatinib et son métabolite actif peuvent être distribués dans le lait maternel. Le rapport lait/plasma des concentrations d'imatinib mesuré chez une patiente était de 0,5 et celui du métabolite était de 0,9, suggérant une distribution plus élevée du métabolite dans le lait. En considérant la concentration de l'imatinib associée à celle de son métabolite et la quantité de lait journalière ingérée par les nourrissons, l'exposition totale attendue devrait être faible (environ 10% de la dose thérapeutique). Cependant, les effets d'une exposition de faible dose chez le nourrisson n'étant pas connus, les femmes traitées par l'imatinib ne doivent pas allaiter.
Fertilité
La fertilité des rats mâles et femelles n'a pas été affectée dans les études précliniques . Aucune étude n'a été effectuée sur des patients recevant de l'imatinib et sur son effet sur la fécondité et la gamétogenèse. Les patients préoccupés par leur fécondité sous traitement par imatinib doivent consulter leur médecin.
L'accès aux informations contenues dans ce Site est ouvert à tous les professionnels de santé.
Les informations données sur le site le sont à titre purement informatif.
Toutefois l'utilisateur exploite les données du site Medzai.net sous sa seule responsabilité : ces informations ne sauraient être utilisées sans vérification préalable par le l’internaute et Medzai.net ne pourra être tenue pour responsable des conséquences directes ou indirectes pouvant résulter de l'utilisation, la consultation et l'interprétation des informations fournies.

 Français
Français Русский
Русский